
| |
Timothy L. Bailey
|
My research seeks to understand all stages of gene expression and to construct computational models of how gene expression is regulated. To accomplish this goal, we develop and employ computational approaches for analyzing genomic, RNA and protein data. These approaches are based on statistical analysis, pattern recognition and mathematical modeling techniques, areas in which we have long experience.
A focus of my work is methods for finding the DNA-binding patterns (motifs) of transcription factor proteins utilizing high-throughput sequencing of chromatin immunoprecipitation data (ChIP-seq). Many of these analysis methods are directly applicable to the study of RNA-binding motifs as well through the analysis of RNA data from "CLIP-seq" and related protocols. Motif analysis is also useful for understanding the function and evolution of proteins.
Making the algorithms and models we develop easily accessible to biologists is a major goal. We try to provide web-based interfaces, and much of my work is included in the popular MEME Suite of sequence analysis tools, which is used by thousands of biologists each month and has been cited over 47,000 times.
| COMBINED MEME SUITE TOOLS: YEARLY and MONTHLY USAGE STATISTICS (all servers) | |
|---|---|
| MEME SUITE UNIQUE USERS | MEME SUITE RUNS |
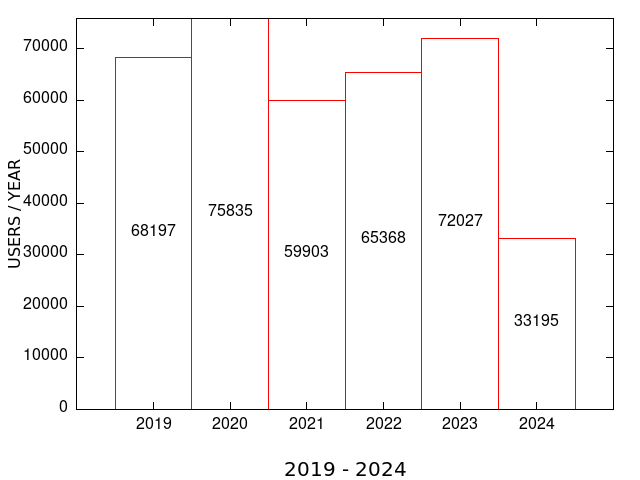 |
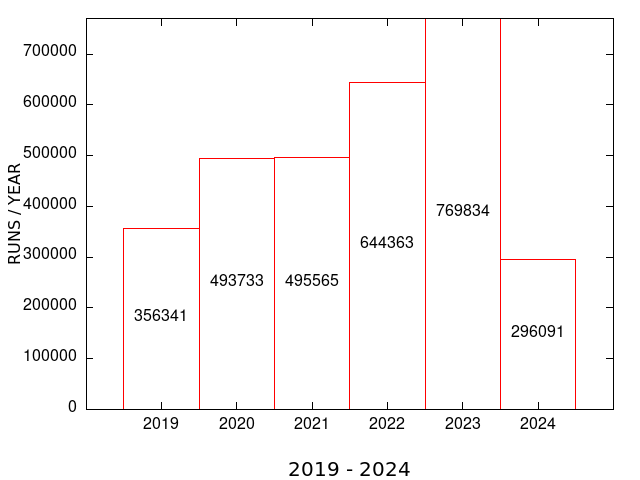 |
| MEME SUITE CPU TIME | MEME SUITE MAX RUNS per USER |
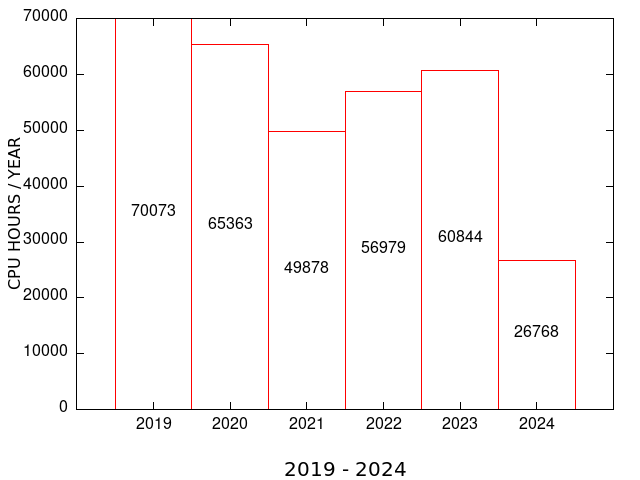 |
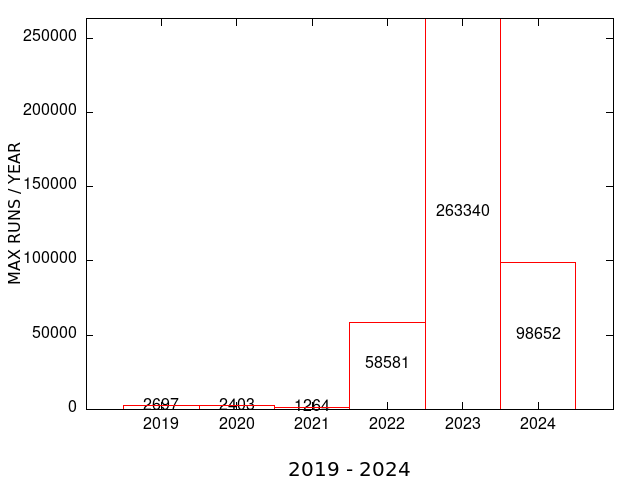 |
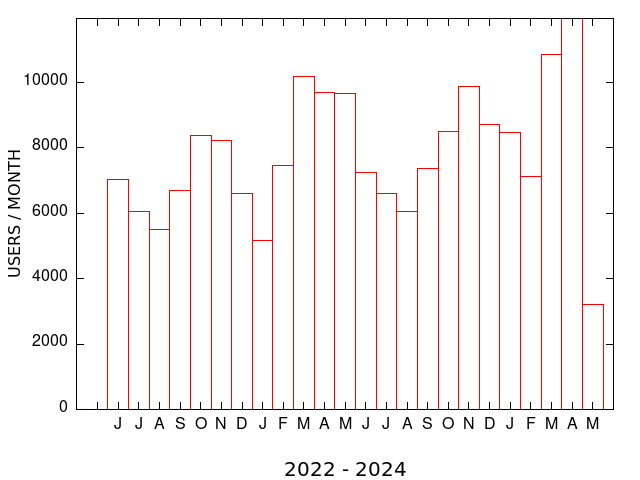
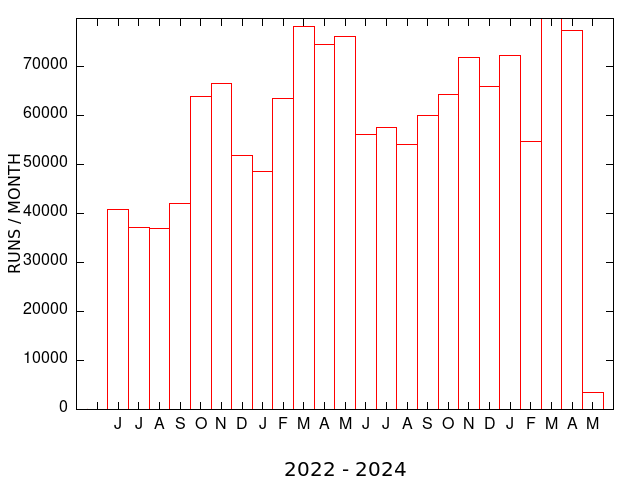
| MEME SUITE UNIQUE USERS (MONTHLY) | MEME SUITE RUNS (MONTHLY) |
 |
 |
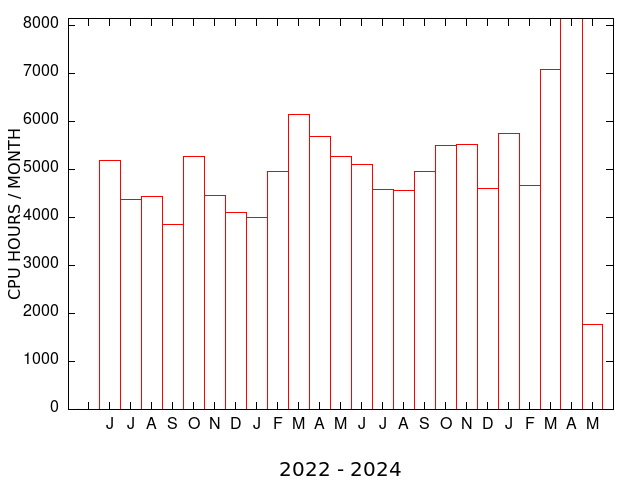
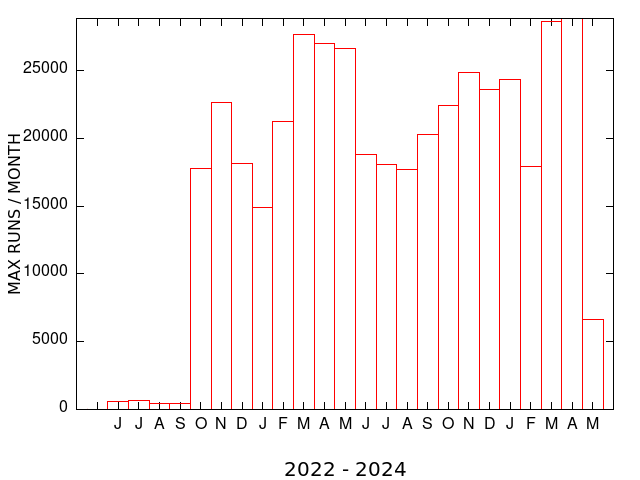
| MEME SUITE CPU TIME (MONTHLY) | MEME SUITE MAX RUNS per USER (MONTHLY) |
 |
 |